DNA–DNA interactions
DNA is one the best-recognized molecules. We are all familiar with the famous double-helix that carries instructions for manufacturing and assembling all the components of a living organism. But DNA is more than just a sequence of letters arranged on rigid ladder rungs; it is a polymer with unusual physical properties that, at times, appear to contradict one another. For example, DNA carries a large negative charge, yet under the right conditions, DNA molecules attract and condense into a compact state. The physical properties of DNA are broadly exploited by cells to perform the molecular feats neccessary for life including storage of information, replication and repair of that information, and regulataion of how that information is expressed. Hence, elucidation of the molecular mechanisms that govern the behavior of DNA in solution comprises a core thrust of our research.
The physical properties of DNA have been suggested to play a central role in spatio-temporal organization of eukaryotic chromosomes. Experimental correlations have been established between the local nucleotide content of DNA and the frequency of inter- and intra-chromosomal contacts but the underlying physical mechanism remains unknown. Here, we combine fluorescence resonance energy transfer (FRET) measurements, precipitation assays, and molecular dynamics simulations to characterize the effect of DNA nucleotide content, sequence, and methylation on inter-DNA association and its correlation with DNA looping. First, we show that the strength of DNA condensation mediated by poly-lysine peptides as a reduced model of histone tails depends on the DNA’s global nucleotide content but also on the local nucleotide sequence, which turns out to be qualitatively same as the condensation by spermine. Next, we show that the presence and spatial arrangement of C5 methyl groups determines the strength of inter-DNA attraction, partially explaining why RNA resists condensation. Interestingly, multi-color single molecule FRET measurements reveal strong anti-correlation between DNA looping and DNA–DNA association, suggesting that a common biophysical mechanism underlies them. We propose that the differential affinity between DNA regions of varying sequence pattern may drive the phase separation of chromatin into chromosomal subdomains.
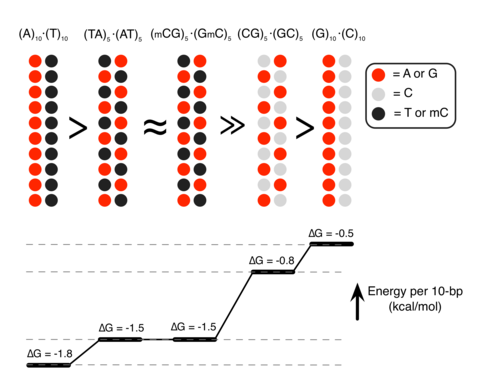
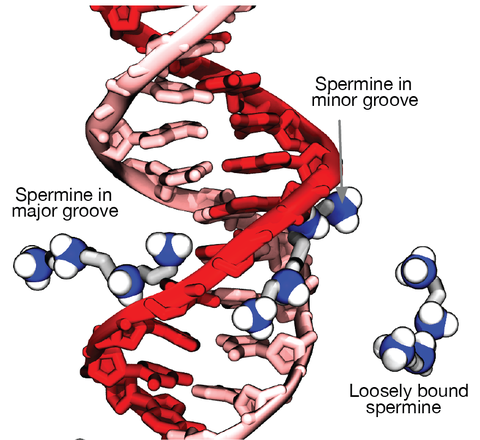
Although proteins mediate highly ordered DNA organization in vivo, theoretical studies suggest that homologous DNA duplexes can preferentially associate with one another even in the absence of proteins. Here we combine molecular dynamics simulations with single-molecule fluorescence resonance energy transfer experiments to examine the interactions between duplex DNA in the presence of spermine, a biological polycation. We find that AT-rich DNA duplexes associate more strongly than GC-rich duplexes, regardless of the sequence homology. Methyl groups of thymine acts as a steric block, relocating spermine from major grooves to interhelical regions, thereby increasing DNA–DNA attraction. Indeed, methylation of cytosines makes attraction between GC-rich DNA as strong as that between AT-rich DNA. Recent genome-wide chromosome organization studies showed that remote contact frequencies are higher for AT-rich and methylated DNA, suggesting that direct DNA–DNA interactions that we report here may play a role in the chromosome organization and gene regulation.
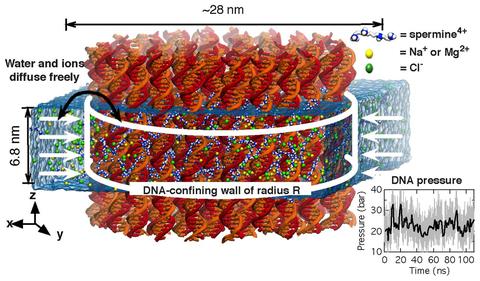
Spontaneous assembly of DNA molecules into compact structures is ubiquitous in biological systems. Experiment has shown that polycations can turn electrostatic self-repulsion of DNA into attraction, yet the physical mechanism of DNA condensation has remained elusive. Here, we report the results of atomistic molecular dynamics simulations that elucidated the microscopic structure of dense DNA assemblies and the physics of interactions that makes such assemblies possible. Reproducing the setup of the DNA condensation experiments, we measured the internal pressure of DNA arrays as a function of the DNA–DNA distance, showing a quantitative agreement between the results of our simulations and the experimental data. Analysis of the MD trajectories determined the DNA–DNA force in a DNA condensate to be pairwise, the DNA condensation to be driven by electrostatics of polycations and not hydration, and the concentration of bridging cations, not adsorbed cations, to determine the magnitude and the sign of the DNA–DNA force. Finally, our simulations quantitatively characterized the orientational correlations of DNA in DNA arrays as well as diffusive motion of DNA and cations.
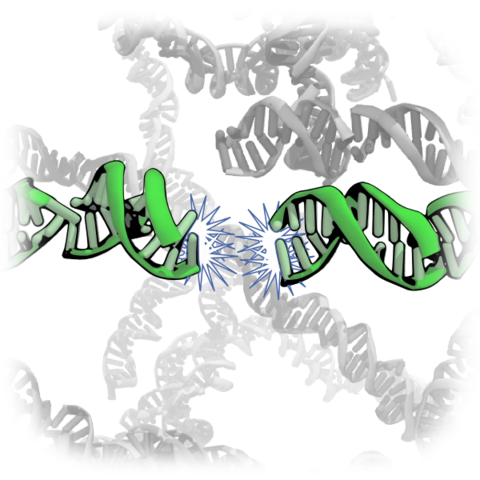
Recent experiments [Nakata, M. et al., End-to-end stacking and liquid crystal condensation of 6 to 20 basepair DNA duplexes. Science 2007; 318:1276-1279] have demonstrated spontaneous end-to-end association of short duplex DNA fragments into long rod-like structures. By means of extensive all-atom molecular dynamic simulations, we characterized end-to-end interactions of duplex DNA, quantitatively describing the forces, free energy and kinetics of the end-to-end association process. We found short DNA duplexes to spontaneously aggregate end-to-end when axially aligned in a small volume of monovalent electrolyte. It was observed that electrostatic repulsion of 5'-phosphoryl groups promoted the formation of aggregates in a conformation similar to the B-form DNA double helix. Application of an external force revealed that rupture of the end-to-end assembly occurs by the shearing of the terminal base pairs. The standard binding free energy and the kinetic rates of end-to-end association and dissociation processes were estimated using two complementary methods: umbrella sampling simulations of two DNA fragments and direct observation of the aggregation process in a system containing 458 DNA fragments. We found the end-to-end force to be short range, attractive, hydrophobic and only weakly dependent on the ion concentration. The relation between the stacking free energy and end-to-end attraction is discussed as well as possible roles of the end-to-end interaction in biological and nanotechnological systems.
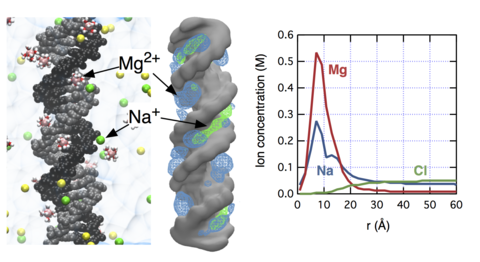
The concept of "ion atmosphere" is prevalent in both theoretical and experimental studies of nucleic acid systems, yet the spatial arrangement and the composition of ions in the ion atmosphere remain elusive, in particular when several ionic species (e.g., Na(+), K(+), and Mg(2+)) compete to neutralize the charge of a nucleic acid polyanion. Complementing the experimental study of Bai and co-workers (J. Am. Chem. Soc.2007, 129, 14981), here we characterize ion atmosphere around double-stranded DNA through all-atom molecular dynamics simulations. We demonstrate that our improved parametrization of the all-atom model can quantitatively reproduce the experimental ion-count data. Our simulations determine the size of the ion atmosphere, the concentration profiles of ionic species competing to neutralize the DNA charge, and the sites of the cations' preferential binding at the surface of double-stranded DNA. We find that the effective size of the ion atmosphere depends on both the bulk concentration and valence of ions: increasing either reduces the size of the atmosphere. Near DNA, the concentration of Mg(2+) is strongly enhanced compared to monovalent cations. Within the DNA grooves, the relative concentrations of cations depend on their bulk values. Nevertheless, the total charge of competing cations buried in the DNA grooves is constant and compensates for about ~30% of the total DNA charge.
DNA-DNA interactions are important for genome compaction and transcription regulation. In studies of such complex processes, DNA is often modeled as a homogeneously charged cylinder and its electrostatic interactions are calculated within the framework of the Poisson-Boltzmann equation. Commonly, a charge adaptation factor is used to address limitations of this theoretical approach. Despite considerable theoretical and experimental efforts, a rigorous quantitative assessment of this parameter is lacking. Here, we comprehensively characterized DNA-DNA interactions in the presence of monovalent ions by analyzing the supercoiling behavior of single DNA molecules held under constant tension. Both a theoretical model and coarse-grained simulations of this process revealed a surprisingly small effective DNA charge of 40% of the nominal charge density, which was additionally supported by all-atom molecular dynamics simulations.
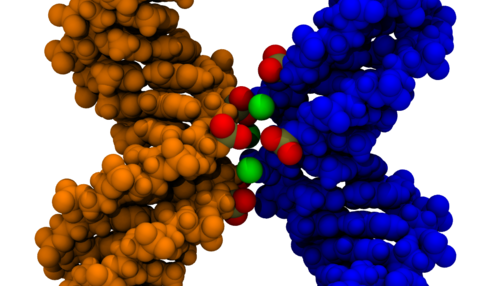
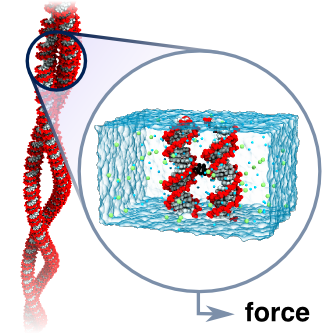
The dependence of the effective force on the distance between two DNA molecules was directly computed from a set of extensive all-atom molecular dynamics simulations. The simulations revealed that in a monovalent electrolyte the effective force is repulsive at short and long distances but can be attractive in the intermediate range. This attractive force is, however, too weak (approximately 5 pN per turn of a DNA helix) to induce DNA condensation in the presence of thermal fluctuations. In divalent electrolytes, DNA molecules were observed to form a bound state, where Mg(2+) ions bridged minor groves of DNA. The effective force in divalent electrolytes was predominantly attractive, reaching a maximum of 42 pN per one turn of a DNA helix.