Atomic Resolution Brownian Dynamics
Download the latest release of ARBD
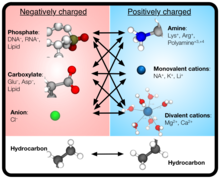
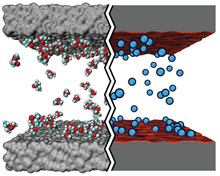
Complementing experimental investigations, computational approaches yield a molecular picture of processes that are too small and fast to resolve experimentally in biological and nanotechnological systems. The most widely employed biological simulation method, all-atom MD, describes molecules with atomic resolution. However, MD is computationally expensive and might be overkill for many tasks. Coarse-graind (CG) Brownian dynamics (BD) simulations are a promising alternative for modeling macromolecular complexes.
BD achieves computational economy while keeping molecular-level detail by modeling the solute through point-like particles with parameterized properties but treating solvent molecules implicitly. Hence, CG BD simulations can be used to overcome the constraints of all-atom MD simulations, which are typically limited by computational resources to time and length scales under about 10 microseconds and 30 nanometers.
Atomic Resolution Brownian Dynamics (ARBD) is a code that takes advantage of GPUs to facilitate fast simulations. Uniquely, ARBD supports models that contain both point-like particles and grid-specified physical particles that possess both position and orientation.
You can download ARBD, compile with a recent version of the CUDA toolkit and begin testing included examples. Please contact Chris Maffeo at cmaffeo2@illinois.edu if you have any trouble using the software. Also, please note that the software is currently offered as an alpha release.