New tricks for old dogs: Improving the accuracy of biomolecular force fields by pair-specific corrections to non-bonded interactions
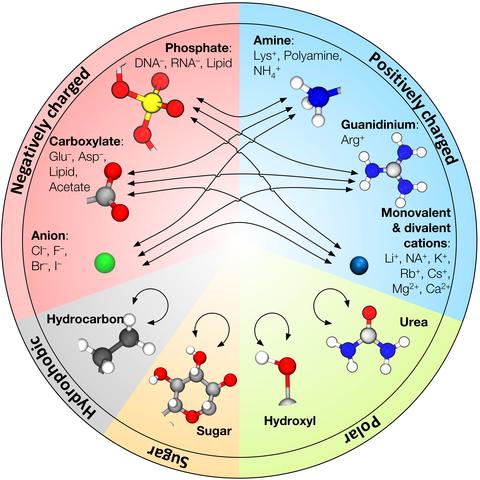
In contrast to ordinary polymers, the vast majority of biological macromolecules adopt highly ordered three-dimensional structures that define their functions. The key to folding of a biopolymer into a unique 3D structure or to assembly of several biopolymers into a functional unit is a delicate balance between the attractive and repulsive forces that also makes such self-assembly reversible under physiological conditions. The all-atom molecular dynamics (MD) method has emerged as a powerful tool for studies of individual biomolecules and their functional assemblies, encompassing systems of ever increasing complexity. However, advances in parallel computing technology have outpaced development of the underlying theoretical models—the molecular force fields, pushing the MD method into untested territory. Recent tests of the MD method have found the most commonly used molecular force fields to be out of balance, overestimating attractive interactions between charged and hydrophobic groups, which can promote artificial aggregation in MD simulations of multi-component protein, nucleic acid, and lipid systems. One route to improving the force fields is through the NBFIX corrections method, in which the intermolecular forces are calibrated against experimentally measured quantities such as osmotic pressure by making atom pair-specific adjustments to the non-bonded interactions. In this article, we review development of the NBFIX corrections to the AMBER and CHARMM force fields and discuss their implications for MD simulations of electrolyte solutions, dense DNA systems, Holliday junctions, protein folding, and lipid bilayer membranes.