In situ structure and dynamics of DNA origami determined through molecular dynamics simulations.
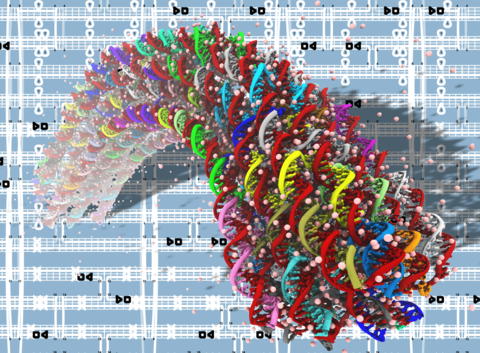
The DNA origami method permits folding of long single-stranded DNA into complex 3D structures with subnanometer precision. Transmission electron microscopy, atomic force microscopy, and recently cryo-EM tomography have been used to characterize the properties of such DNA origami objects, however their microscopic structures and dynamics have remained unknown. Here, we report the results of all-atom molecular dynamics simulations that characterized the structural and mechanical properties of DNA origami objects in unprecedented microscopic detail. When simulated in an aqueous environment, the structures of DNA origami objects depart from their idealized targets as a result of steric, electrostatic, and solvent-mediated forces. Whereas the global structural features of such relaxed conformations conform to the target designs, local deformations are abundant and vary in magnitude along the structures. In contrast to their free-solution conformation, the Holliday junctions in the DNA origami structures adopt a left-handed antiparallel conformation. We find the DNA origami structures undergo considerable temporal fluctuations on both local and global scales. Analysis of such structural fluctuations reveals the local mechanical properties of the DNA origami objects. The lattice type of the structures considerably affects global mechanical properties such as bending rigidity. Our study demonstrates the potential of all-atom molecular dynamics simulations to play a considerable role in future development of the DNA origami field by providing accurate, quantitative assessment of local and global structural and mechanical properties of DNA origami objects.
These animations illustrate the dynamic nature of a DNA origami structure. Each simulation shows the conformation of a DNA layer in a three-layer honeycomb structure observed during 130 nanoseconds of molecular dynamics. The backbone and bases of DNA are shown as tubes; each DNA strand is show using a unique color. Basepairs from the non-peripheral region of the structure that remained broken for 10 nanoseconds or more are highlighted using the vdW (spheres) representations. In frame zero, the circle indicates a basepair that was observed to break and reform during the simulation.
This animation illustrates equilibrium fluctuations of a three-layer honeycomb structure observed during a 130 ns molecular dynamics simulation. The DNA origami structure is shown using a color-coded chicken wire representation. The wireframe (black lines) connects the centers of mass of the DNA basepairs that form continuous double-stranded DNA cylinders in the original DNA origami design. The lines between the centers of mass of the same-index basepairs connect the wireframe. The length of the lines indicate the local inter-DNA distance, which is color-coded from blue (short distance) to red (long distance).
This animation illustrates a 120 ns molecular dynamics simulation of a square-lattice DNA origami structure. The DNA origami structure is shown using a color-coded chicken wire representation. Individual animations illustrate the dynamics of the same structure viewed from four different sides.
Equilibrium fluctuations of a honeycomb structure featuring a 90-degree programmable bend. The animations covers 30 ns of a molecular dynamics simulations; the simulation system contains over 3 million atoms. The DNA origami structure is shown using a color-coded chicken wire representation; water and ions are not shown. During the simulation, the local structure of DNA origami undergoes considerable fluctation; the bend angle of the structure remains close to 90 degrees.
This animation illustrates a simulation protocal that was used to produce a 90-degree bend in a honeycomb structure. Initial all-atom structure constructed by converting the caDNAno design to all-atom coordinates. During the first 2 ns of equilibration, all non-hydrogen atoms of DNA bases were restrained to their initial coordinates; magnesium ions migrated to their equilibrium locations within the DNA origami structure. A bend in the structure was produced by allowing the DNA helices to expand and shrink according to the insertions and deletions introduced in the design. To preserve the local order of the structure, the distance between non-hydrogen atoms of the DNA bases within 10 Å was restrained. Such elastic-network-like restraints were realized by introducing harmonic bonds using the extra bonds feature of NAMD. The restraints were removed in two steps producing the programmed 90 degree bend in the absence of the restraints.