Jejoong Yoo

Department of Physics
Jejoong Yoo received his B.S. degree in physics and molecular biology from the Seoul National University in 2001, and his Ph.D. in biophysics from the University of Wisconsin-Madison in 2010. He is currently a postdoctoral fellow working with Professor Aksimentiev and Professor Ha at CPLC.
For complete publication list, download CV | Google Scholar | ResearchGate | ResearchID | Pubmed
Research highlights:
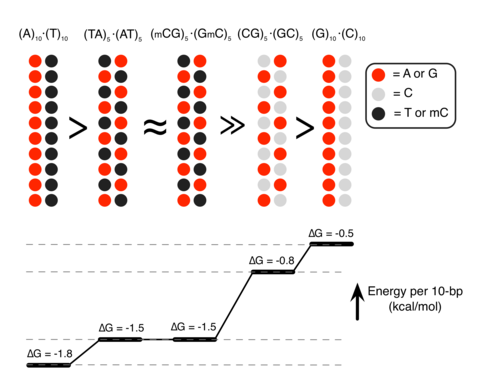
Epigenetic control of DNA condensation
See Nature Communications 2016. DNA condensation is important for gene regulations. In this paper, we demonstrate that the polyamine-mediated DNA condensation can be controlled by the sequence and methylation of DNA.
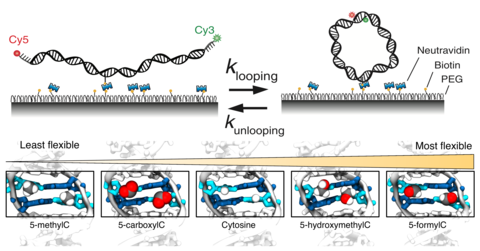
Epigenetic control of DNA flexibility
See Nature Communicatgions 2016. DNA flexibility is an important materials property of DNA that can control the stability of nucleosomal DNA. In this paper, we demonstrate how various epigenetic modifications of DNA modulate the DNA flexibility.
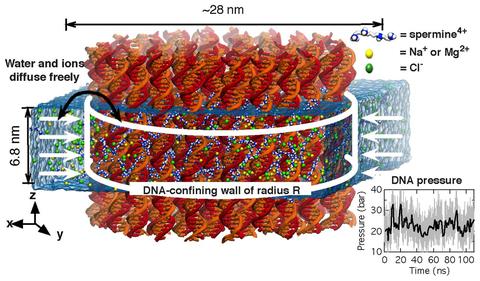
Physics of DNA condensation
See Nucleic Acids Research 2016. Polyamine-mediated DNA condensation is an interesting research area in soft matter physics showing interesting phase behaviors. Yet, application of MD simulation to the DNA condensation was limited due to imperfection of the force fields. Using AMBER force field in combination with our CUFIX corrections, we quantitatively demonstrate the DNA condensation phenomena at an atomstic resolution.
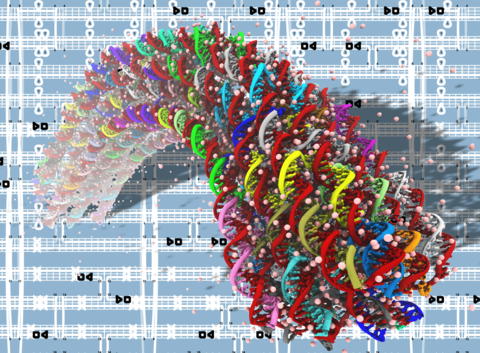
DNA origami nanotechnology
See PNAS 2013 & Movies. This is THE FIRST MD simulation study of DNA origami. We show that MD is an effective method to characterize the mechanical properties of DNA origami.
See Nucleic Acids Research 2016. De novo structure prediction of DNA origami using MD simulations.
See ACS Nano 2015. The first MD study on electrical properties of DNA origami in collaboration with the Keyser group at U Cambridge.
See New J of Physics 2016. The first MD study of DNA brick.
DNA channels
See JPCL 2015. The FIRST MD study of biomimetic DNA channel characterizing its electrical, mechanical, and transport properties.
See Nano Lett 2016 | ACS Nano 2016. Exploration of various DNA-based channels in collaboration with the Keyser group at U Cambridge.

Champaign-Urbana nbFIX (CUFIX) for AMBER and CHARMM force fields
MD force fiels such as AMBER and CHARMM have demonstrated a great potential over decades. But, their applications to inter-molecular interactions (e.g., protein-DNA interactions) are limited due to imperfection of parameters. For example, the standard AMBER and CHARMM predict a wrong sign for lysine-peptide-mediated DNA-DNA interaction forces (See JCTC 2016)! In a series of publications, we demonstrated that simple corrections of Lennard-Jones parameters can dramatically improve the realism of inter-molecular forces.
Download CUFIX for Gromacs, NAMD, CHARMM, and ANTON.
See JCTC 2016 on inter-molecular forces among nucleic acids, proteins, and lipid.
See JPCL 2012. CUFIX for ions such as Na, K, Li, and Mg.
See Biopolymers 2016. CUFIX for calcium ion.

Protein folding
See JPCL 2016. The MD community generally agree that the standard force fields such as AMBER and CHARMM predict overly condensed denatured conformation of proteins. This shortcoming is an important barrier that we have to overcome for realistic simulations of protein folding process or conformational sampling of intrinsically disordered proteins. In this paper, we demonstrate that both standard AMBER & CHARMM overestimate the charge-charge and hydrophobic interactions, resulting in too compact denatured conformations. Then, we demonstrate that the folding kinetics and the denatured ensemble in the REMD simulations of WW domain and villin head piece proteins can be dramatically improved by using CUFIX.

Nanopore sequencing
See ACS Nano 2016. Biological nanopore such as MspA is a promising candidate for nanopore sequencing. In this paper, we demonstrate the role of water in determining the ionic current blockade by ssDNA using the ANTON supercompter.

Publications
-
"Improved model of hydrated calcium ion for molecular dynamics simulations using classical biomolecular force fields." Biopolymers 105:752-763 (2016).
 bip22868-sup-0001-suppinfo01.pdf (921.41 KB)
bip22868-sup-0001-suppinfo01.pdf (921.41 KB) - "De Novo Reconstruction of DNA Origami Structures through Atomistic Molecular Dynamics Simulation." Nucleic Acids Research 44:3013-3019 (2016).
-
"Direct evidence for sequence-dependent attraction between double-stranded DNA controlled by methylation." Nature Communications 7:11045 (2016). ncomms11045-s1.pdf (1.81 MB)
-
"Effect of Cytosine Modifications on DNA Flexibility and Nucleosome Mechanical Stability." Nature Communications 7:10813 (2016). ncomms10813-s1.pdf (529.11 KB)
-
"The structure and intermolecular forces of DNA condensates." Nucleic Acids Research 44:2036-2046 (2016). dnapack_supp.pdf (9.65 MB)
-
"Improved Parameterization of Amine–Carboxylate and Amine–Phosphate Interactions for Molecular Dynamics Simulations Using the CHARMM and AMBER Force Fields." Journal of Chemical Theory and Computation 12:430-443 (2016). ct5b00967_si_001.pdf (2.12 MB)
-
"Molecular Dynamics of Membrane-Spanning DNA Channels: Conductance Mechanism, Electro-Osmotic Transport, and Mechanical Gating." The Journal of Physical Chemistry Letters 6:4680-4687 (2015). Supporting Information (8.97 MB)
-
"Ionic Conductivity, Structural Deformation, and Programmable Anisotropy of DNA Origami in Electric Field." ACS Nano 9:1420-1433 (2015). Supporting Information (8.81 MB)
- "Close encounters with DNA." J Phys Condens Matter 26:413101 (2014).
-
"In situ structure and dynamics of DNA origami determined through molecular dynamics simulations." Proc Natl Acad Sci U S A 110:20099-104 (2013). Supporting Information (4.15 MB)
- "Modeling and simulation of ion channels." Chem Rev 112:6250-6284 (2012).
-
"Competitive binding of cations to duplex DNA revealed through molecular dynamics simulations." J Phys Chem B 116:12946-54 (2012). jp306598y_si_001.pdf (343.44 KB)
-
"Improved Parametrization of Li+, Na+, K+, and Mg2+ Ions for All-Atom Molecular Dynamics Simulations of Nucleic Acid Systems." The Journal of Physical Chemistry Letters 3:45-50 (2012). jz201501a_si_001.pdf (1.69 MB)
-
"Refined Parameterization of Nonbonded Interactions Improves Conformational Sampling and Kinetics of Protein Folding Simulations." Journal of Physical Chemistry Letters 7:3812-3818 (2016). jz6b01747_si_001.pdf (6.89 MB)
-
"Large-Conductance Transmembrane Porin Made from DNA Origami." ACS Nano 10:8207-8214 (2016). supporting_information.pdf (3.19 MB)
-
"Ion Channels Made from a Single Membrane-Spanning DNA Duplex." Nano Letters 16:4665-4669 (2016). supporting_information.pdf (1.32 MB)
-
"Molecular mechanics of DNA bricks: in situ structure, mechanical properties and ionic conductivity." New Journal of Physics 18:055012 (2016). supporting_information.pdf (7.84 MB)