Grid-steered molecular dynamics
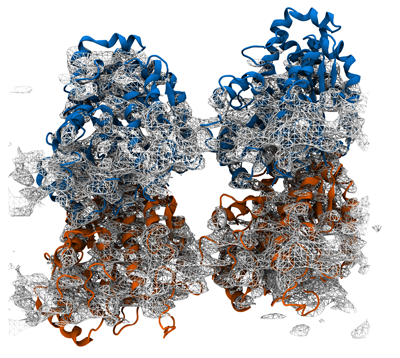
Grid-steered molecular dynamics (G-SMD) is a flexible method for applying forces that our group has implemented in NAMD. Much as in experiment, simulation studies often involve perturbing the system in some way and monitoring the result. As simulations have become bigger, longer, and more complex, the need for more sophisticated forcing techniques has increased. In the G-SMD method, an arbitrary external potential field, defined on a grid, is applied to desired target atoms with arbitrary coupling, making it a very flexible tool. Because it is coded natively in NAMD, the performance impact of G-SMD is minimal. G-SMD provides tremendous flexibility in the forces that can be applied. It was originally designed to apply an enhanced electrostatic field to DNA threaded through the membrane protein α-hemolysin in order to realistically increase the DNA translocation speed. It is the basis for a method of combining crystallographic structures and cryo-EM maps to obtain an all-atom model developed by Trabuco et al. and called Molecular Dynamics Flexible Fitting (MDFF). A variation of this approach was also recently used by our group to study the mechanical properties of a complete microtubule.

