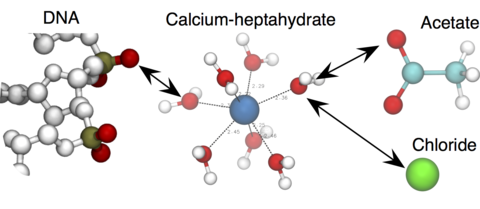
Calcium ions (Ca2+) play key roles in various fundamental biological processes such as cell signaling and brain function . Molecular dynamics (MD) simulations have been used to study such interactions, however, the accuracy of the Ca2+ models provided by the standard M D force fields has not been rigorously tested. Here, we assess the performance of the Ca2+ models from the most popular classical force fields AMBER and CHARMM by computing the osmotic pressure of model compounds and the free energy of DNA–DNA inter act i on s. In the simulations performed using the two standard models, Ca2+ ions are seen to form artificial clusters with chloride, acetate, and phosphate species; the osmotic pressure of CaAc2and CaCl2solutions is a small fraction of the experimental values for both force fields. Using the standard parameterization of Ca2+ ions in the simulations of Ca2+-mediated DNA-DNA interact ions leads to qualitatively wrong outcomes: both AMBER and CHARMM simulations suggest strong inter-DNA attraction whereas, in experiment, DNA molecules repel one another. The artificial attraction of Ca2+to DNA phosphate is strong enough to affect the direction of the electric field-driven translocation of DNA through a solid-state nanopore. To address these shortcomings of the standard Ca2+ model, we introduce a custom model of a hydrated Ca2+ ion and show that using our model brings the results of the above MD simulations in quantitative agreement with experiment. Our improved model of Ca2+ can be readily applied to MD simulations of various biomolecular systems, including nucleic acids, proteins and lipid bilayer membranes.