CUFIX: Non-bonded Fix (NBFIX) parameters for the CHARMM and AMBER force fields
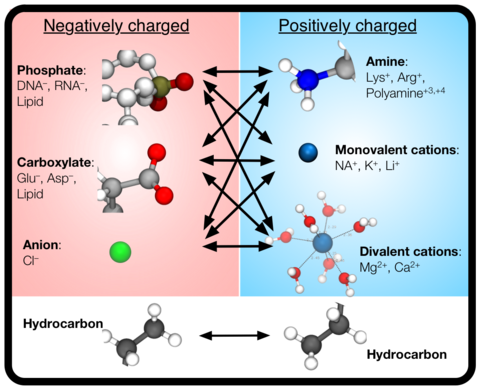
Over the past decades, molecular dynamics (MD) simulations of biomolecules have become a mainstream biophysics technique. As the length and time scales amenable to the MD method increase, shortcomings of the empirical force fields—which were developed and validated using relatively short simulations of small molecules—become apparent. One common artifact is artificial aggregation of water-soluble biomolecules, which has been observed in a variety of systems, including electrolyte solutions, intrinsically disordered proteins, lipid bilayer membranes and DNA arrays. Here, we report a systematic refinement of Lennard-Jones parameters (CUFIX) describing amine-carboxyate, amine-phosphate, and aliphatic carbon-carbon interactions, which brings the results of MD simulations of proteins, nucleic acids, and lipids in remarkable agreement with experiments. To refine the amine-carboxylate and aliphatic carbon-carbon interactions, we matched the simulated osmotic pressure of amino acid solutions to the experimental data. Similarly, we refined the amine-phosphate interaction by matching the simulated and experimental osmotic pressure of a DNA array. We demonstrate the utility of our CUFIX corrections through simulations of lysine-mediated DNA—DNA forces, lipid-bilayer membranes and folded proteins. As our refinement neither affects the existing parameterization of bonded interaction nor does it alter the solvation free energies, it improves realism of an MD simulation without introducing any new artifacts.
How to use CUFIX (contact Jejoong Yoo jejoong@gmail.com):
-
AMBER ff99/ff14 variants in the Gromacs format:
- > Option 1: If you want to use a complete package that was used for our publications, download one of the followings. Make sure our CUFIX corrections are optimized with the TIP3P water model (use tip3p.itp).
-
> Option 2: If you want to integrate CUFIX to your version of AMBER ff99/ff14 variants, follow these steps.
- >> Download ff99sb-ildn-phi-bsc0-cufix package.
- >> Copy the following files in the downloaded package to your gromacs-format ff99 folder: cufix.itp, mg-sol6.itp mg-sol6.pdb ca-sol7.itp ca-sol7.pdb.
- >> Replace atom types of O1P and O2P atoms (O2) with ON2 for all nucleotides in dna.rtp and rna.rtp. ON2 atom type is defined in cufix.itp.
- >> Add #include "cufix.itp" to forcefield.itp between #include "ffnonbonded.itp" and #include "ffbonded.it".
- >> Delete the following ions from ffnonbonded.itp: Li, Na, K, Cl, MG, Rb, Cs, F, Br, I. CUFIX uses new ion parameters by the Cheatham group.
- >> You're ready to go! Make sure that CUFIX is optimized with tip3p.itp.
-
AMBER ff99/ff14 variants in AMBER format
- > Download cufix.tar and untar in the amber16/dat/leap directory.
- > For DNA and RNA, we prepared leaprc.DNA.bsc1.cufix and leaprc.RNA.OL3.cufix. In these files, we introduce ON2 atom type for the phosphate oxygen atoms to differentiate them from the carboxylate atom type O2.frcmod.ff99cufix file will work with any ff99 and ff14 variant protein force field in combination with leaprc.water.tip3p.
- > For example, do the followings in the leap command:
source <A FF99 PROTEIN FORCE FIELD CMD FILE>
source leaprc.water.tip3p
cufix = loadamberparams frcmod.ff99cufix
-
CHARMM 36/27/22 force fields
- > Download CHARMM36/27/22 force fields: stream file for NAMD packages
- > Replace the standard toppar_water_ions.str with the downloaded file.
- > Our CUFIX corrections are optimized for CHARMM36-version ion parameters (LIT, SOD, K, MG, CAL and CL) with CHARMM-format TIP3P water model.
- > For magnesium and calcium ions, we use hexa- or hepta-hydrated forms, respectively. The downloaded file contains RESI definitions for these Mg/Ca-water complexes, but additional bonds (extrabonds) are required between Mg/Ca and water oxygen atoms. Please refer to our DNA origami tutorial to learn how to use it.
- Anton
- > Download amber99SBstar-ILDN-bsc0-nbfix in VIPARR format.
- > Just use it the same way as the other force fields at $VIPARR3_FFDIR.
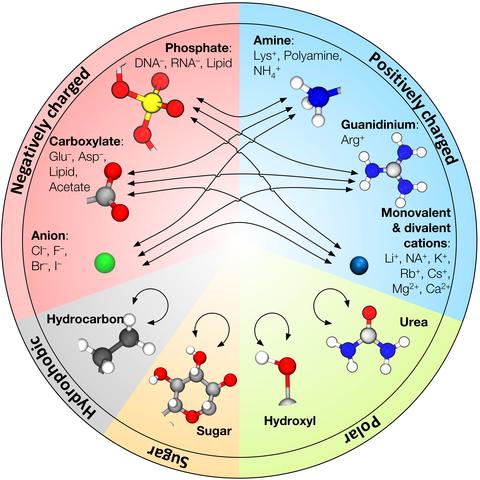
In contrast to ordinary polymers, the vast majority of biological macromolecules adopt highly ordered three-dimensional structures that define their functions. The key to folding of a biopolymer into a unique 3D structure or to assembly of several biopolymers into a functional unit is a delicate balance between the attractive and repulsive forces that also makes such self-assembly reversible under physiological conditions. The all-atom molecular dynamics (MD) method has emerged as a powerful tool for studies of individual biomolecules and their functional assemblies, encompassing systems of ever increasing complexity. However, advances in parallel computing technology have outpaced development of the underlying theoretical models—the molecular force fields, pushing the MD method into untested territory. Recent tests of the MD method have found the most commonly used molecular force fields to be out of balance, overestimating attractive interactions between charged and hydrophobic groups, which can promote artificial aggregation in MD simulations of multi-component protein, nucleic acid, and lipid systems. One route to improving the force fields is through the NBFIX corrections method, in which the intermolecular forces are calibrated against experimentally measured quantities such as osmotic pressure by making atom pair-specific adjustments to the non-bonded interactions. In this article, we review development of the NBFIX corrections to the AMBER and CHARMM force fields and discuss their implications for MD simulations of electrolyte solutions, dense DNA systems, Holliday junctions, protein folding, and lipid bilayer membranes.
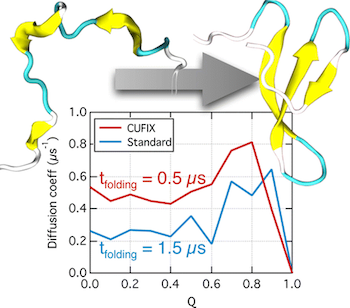
Recent advances in computational technology have enabled brute-force molecular dynamics (MD) simulations of protein folding using physics-based molecular force fields. The extensive sampling of protein conformations afforded by such simulations revealed, however, considerable compaction of the protein conformations in the unfolded state, which is inconsistent with experiment. Here, we show that a set of surgical corrections to nonbonded interactions between amine nitrogen–carboxylate oxygen and aliphatic carbon–carbon atom pairs can considerably improve the realism of protein folding simulations. Specifically, we show that employing our corrections in ∼500 μs all-atom replica-exchange MD simulations of the WW domain and villin head piece proteins increases the size of the denatured proteins’ conformations and does not destabilize the native conformations of the proteins. In addition to making the folded conformations a global minimum of the respective free energy landscapes at room temperature, our corrections also make the free energy landscape smoother, considerably accelerating the folding kinetics and, hence, reducing the computational expense of a protein folding simulation.
Over the past decades, molecular dynamics (MD) simulations of biomolecules have become a mainstream biophysics technique. As the length and time scales amenable to the MD method increase, shortcomings of the empirical force fields, which have been developed and validated using relatively short simulations of small molecules, become apparent. One common artifact is aggregation of water-soluble biomolecules driven by artificially strong charge–charge interactions. Here, we report a systematic atom pair-specific refinement of Lennard-Jones parameters (NBFIX) describing amine–carboxylate and amine–phosphate interactions, which bring MD simulations of basic peptide-mediated nucleic acid assemblies and lipid bilayer membranes into better agreement with experimental data. As our refinement does not affect the existing parametrization of bonded interactions or alter the solvation free energies, it improves the realism of an MD simulation without introducing additional artifacts.
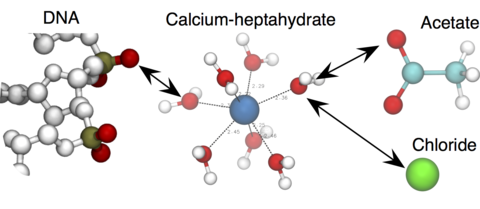
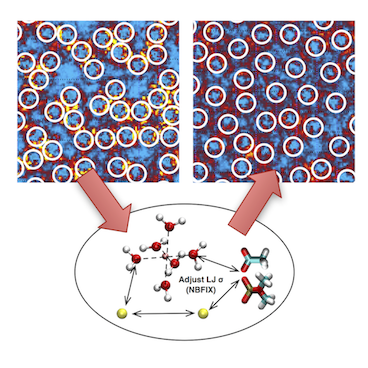
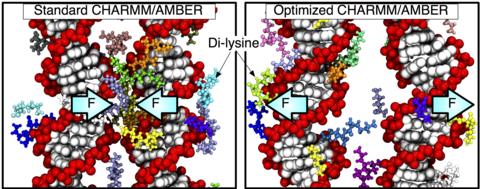
Atomic-scale modeling of compacted nucleic acids has the ability to reveal the inner workings of spectacular biomolecular machines, yet the outcome of such modeling efforts sensitively depends on the accuracy of the underlying computational models. Our molecular dynamics simulations of an array of 64 parallel duplex DNA revealed considerable artifacts of cation−DNA phosphate interactions in CHARMM and AMBER parameter sets: both the DNA arrangement and the pressure inside the DNA arrays were found to be in considerable disagreement with experiment. To improve the models, we fine-tuned van der Waals interaction parameters for specific ion pairs to reproduce experimental osmotic pressure of binary electrolyte solutions of biologically relevant ions. Repeating the DNA array simulations using our parameters produced results consistent with experiment. Our improved parametrization can be directly applied to molecular dynamics simulations of various charged biomolecular systems, including nucleic acids, proteins, and lipid bilayer membranes.